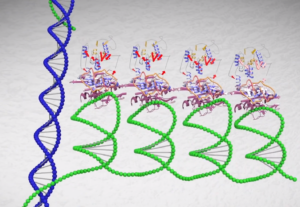
slncRNAs: synthetic long non-coding RNA scaffolds
 |
Triplex-seq: deciphering the rules of DNA triplex formation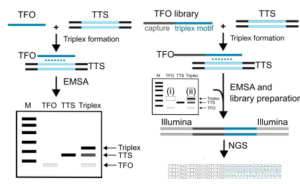
|
UNILIB: a universal library for promoters |
sRNPs: synthetic RNA-protein granules

|
MRG-GRammar:
Massive reverse genomics to decipher gene regulatory grammar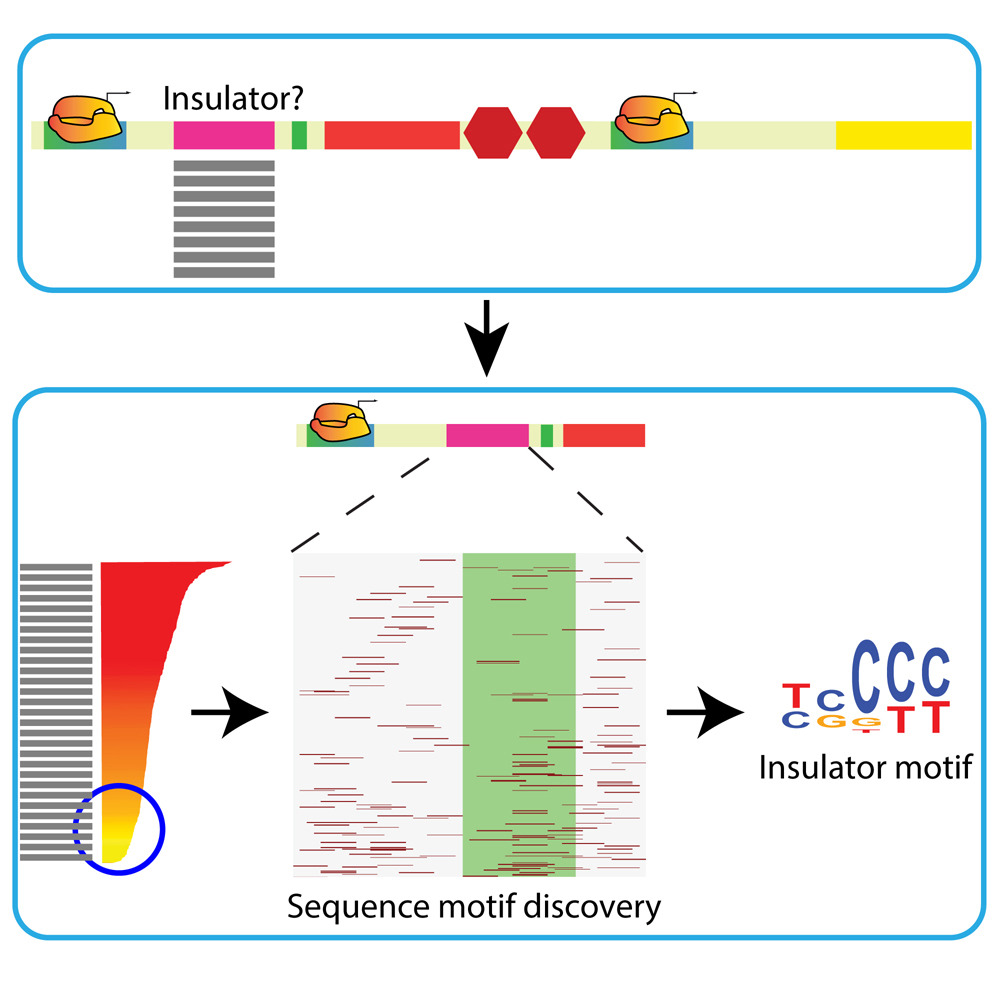 |
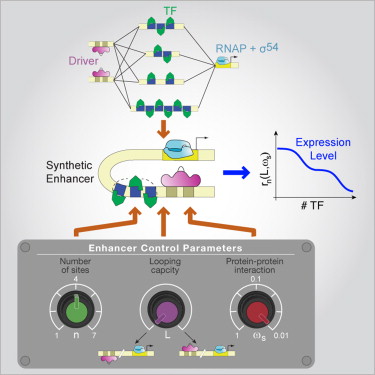
Synthetic enhancers
|